Cystic Fibrosis
AbdulSamad Olagunju / July 05, 2021
11 min read
Welcome to another awesome blog post! This is a term paper I recently wrote about cystic fibrosis.
Quote of the Post:
"When you take charge of your life, there is no longer need to ask permission of other people or society at large. When you ask permission, you give someone veto power over your life." - Geoffrey F. Abert
Introduction
Hacking coughs. Wheezing. Shortness of Breath. These are some of the debilitating symptoms that affect those who suffer from Cystic Fibrosis (CF). In CF, patients struggle with extremely thick mucus in their lungs that is difficult to expel. Moreover, their lungs are extremely susceptible to infection (3).
Imagine the struggles faced by those with this condition. They constantly remain vigilant, knowing an errant cough from a friend might mean days spent in the hospital. Daily, they must painstakingly select the right medications so they can maintain good health. This is a difficult ordeal for any person to manage.
CF occurs due to mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene on chromosome 7 (3). It leads to progressive lung damage and wages war against the pancreas, lungs, and the intestine. Nevertheless, researchers have come up with a myriad of ways to support those with this disorder. In this paper, the pathophysiology of CF will be briefly outlined in addition to the therapies that have given a modicum of control back to the 70,000 worldwide who fight this disease.
The Cystic Fibrosis Transmembrane Conductance Regulator
An autosomal recessive disorder, CF arises due to mutations to the CFTR gene. A functional CFTR gene produces the CFTR protein. CFTR (shown in figure 2) is an essential protein in the human body found on the apical side of epithelial cell membranes in the lungs (10). Its primary purpose is to conduct the transport of chloride ions from the intracellular space of epithelial cells to the extracellular space (5).
According to Rey et al. (11), CFTR belongs to the family of ATP-binding cassette (ABC) proteins and is regulated by Protein Kinase A via cAMP-dependent phosphorylation. The CFTR protein has domains that interact with ATP, and this binding of ATP allows the CFTR channel to open so chloride can be moved across the epithelial cell membrane. For the channel to close, ATP hydrolysis takes place.
Moreover, the CFTR protein also has regions responsible for anchoring it to the cell membrane (5). There are positively charged regions dispersed throughout the membrane-spanning domains of the CFTR protein that allow for high specificity to the chloride anion (11). CFTR also downregulates the epithelial sodium channel and transports bicarbonate ions and other proteins such as glutathione (10).
There are over 2000 mutations pertaining to the CFTR gene that scientists have discovered so far (5). These mutations can affect the function or amount of CFTR protein produced by epithelial cells (3). There are several classes of mutations and these are outlined in figure 1. The most common mutation (70% of patients) is a deletion of Phenylalanine on position 508 of the CFTR protein (5). In particular, the Phenylalanine deletion causes instability to a domain of the CFTR protein that interacts with ATP (5). Consequently, the CFTR protein does not mature and is not transferred to the epithelial cell membrane, and a lack of chloride transportation ensues (1).
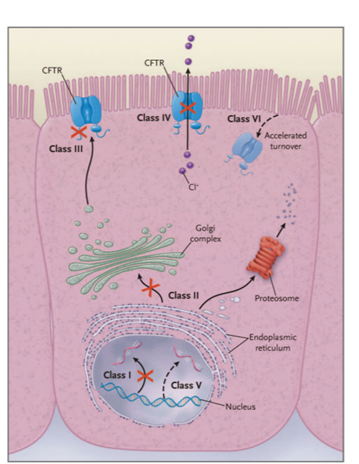
Figure 1- Different classes of CF mutations.
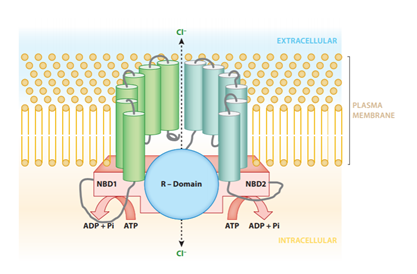
Figure 2- CFTR protein molecular structure.
Consequences to Respiratory Function
Due to a defective CFTR protein, CF patients are extremely susceptible to lung infection. There are two major hypotheses for why this occurs, the “low volume” hypothesis and the “high salt” hypothesis (9).
In the “low volume” hypothesis, CFTR is not functional or present in adequate amounts on epithelial cell membranes. This causes intracellular chloride concentrations to increase, resulting in an osmotic gradient causing water molecules to move into epithelial cells. This results in a lack of water on the surface of epithelial cells. Furthermore, without CFTR to inhibit epithelial sodium ion channels, excess sodium ion and water absorption occurs through epithelial sodium ion channels (10). This results in mucus that is extremely dehydrated and sticky, and less mucus than is necessary to protect the airways from bacteria. The cilia struggle to move properly in this thick mucus and collapse, and the mucus-ciliary elevator does not perform its purpose of protecting the lungs from infection (10). The impeded ciliary activity results in multiple pathogens colonizing the lungs, including bacteria such as Pseudomonas aeruginosa, viruses and fungi (3). This results in an extreme inflammatory response. Neutrophils respond to the pathogens in the lungs by releasing inflammatory chemicals (10). There is structural tissue damage to the airways of a patient with cystic fibrosis, acquired and innate dysregulation of the immune system, and this results in impaired lung function until death (2).
The alternative hypothesis (“high salt hypothesis”) proposed by researchers is that mutant CFTR proteins allow salt levels to rise in airway surface liquid. The increased concentrations of salt in the CF respiratory epithelium result in the inhibition of salt-sensitive antimicrobial peptides called defensins (10). CF mucus has also been found to have low concentrations of a molecule called mucin which helps to clear away bacteria in the airways (10). The high salt concentrations inhibit antibacterial molecules, resulting in recurrent lung infections. Moreover, it has been proposed that a non-functional CFTR protein cannot perform its function as a receptor for P. aeruginosa and mediate the uptake and destruction of this bacteria (10). Patients with CF struggle with obstructive airway disease, chronic lung infections, and bronchiectasis (enlarged parts of lungs resulting in painful coughing) (3). They may also struggle with sinusitis and nasal polyposis (growths on inside of nose) (3). This combination of chronic mucus depletion, inflammation, and infection ravages the lungs and leads to its ruination.
Therapies for CF
For the past 50 years, researchers have discovered new ways to improve the quality of life for those with cystic fibrosis. Those with CF often require methods that can clear up their airways by removing inspissated secretions such as a CF vest (3). Nebulisations that lessen mucus thickness and nebulised antibiotics are often a means of therapy.
According to Castellani and Assael (3), lung transplantation may be required if the lungs display respiratory insufficiency. Furthermore, patients with CF often require anti-inflammatory drugs to combat their heightened inflammatory response. Inflammatory processes often start early in the lungs of an individual with CF. This inflammation is relentless, leading to bronchiectasis (2).
There are two conventional methods for reducing inflammation: corticosteroids and ibuprofen. Ibuprofen is often the better choice, as it has less adverse long-term effects than corticosteroids (2). These options only slow down the progression of lung disease, they are not curative. Gene therapy has shown some promise; however, gene therapy has been limited due to inflammatory reactions and the low efficiency of vectors (carriers that deliver the gene) (3). The complexity of the lung has surprised researchers, and more work is needed in order to fully understand the disease (4). CFTR modulators have had success, with the drug ivacaftor improving the activity of the CFTR channel (8). This results in better functioning of the pancreas, less pathogenic infection in the lungs, and better nutrition for up to four years for patients with CF (3).
A non-functional CFTR protein results in a buildup of thick mucus in the epithelial airway surface. This mucosal obstruction disrupts ciliary movement, resulting in infection. To combat this, researchers studied the effects of inhaled hypertonic saline (HS) on lung function. The rationale for this chemical was that HS should create an osmotic pressure that induces the flow of water back into airway surfaces and consequently, the airway surface liquid should be restored to proper mucus viscosity. According to Boucher (1), Donaldson et al. measured the effect of inhaled 7% HS on lung function. This produced an increase in the basal muco-ciliary clearance after 2 weeks of therapy, and an increase in the forced expiratory volume of CF patients. This data is shown on figure 3. Additionally, a study by Elkins et al. illustrated that inhaling 7% HS four times a day resulted in an increase in lung function over one year and no increase in bacterial densities (1). Research has demonstrated hypertonic saline can be used therapeutically for CF (1).
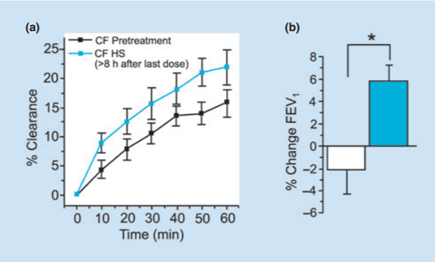
Figure 3- 2 Graphs showing effects of hypertonic saline on CF patient’s FEV1 and %Clearance after a 2-week period of therapy.
Conclusion
CF remains the most life-limiting autosomal recessive condition for Caucasians. When the CFTR gene is mutated, the CFTR protein is either non-functional or unstable. This results in thick mucus in the airways of the respiratory system, and an inability of the cilia to stop pathogens from colonizing sensitive lung tissue. A persistent cycle of infection and inflammation arises, eventually causing respiratory failure and death.
Therapies to attenuate the suffering of CF patients include CFTR modulators, nebulisations, hypertonic saline inhalations, anti-inflammatory drugs, and the CF vest. With genetic therapy continuing to advance by leaps and bounds, it is becoming more evident that there is a chance for researchers to cure this disease by either modifying the CFTR protein, or the CFTR gene. All in all, this may become the most effective way to combat cystic fibrosis in the future.
References
- Boucher RC. Evidence for airway surface dehydration as the initiating event in CF airway disease. Journal of Internal Medicine 261: 5-16, 2007.
- Cantin AM, Hartl D, Konstan MW, and Chmiel JF. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. Journal of Cystic Fibrosis 14: 419-430, 2015.
- Castellani C, and Assael BM. Cystic fibrosis: a clinical view. Cellular and Molecular Life Sciences 74: 129-140, 2016.
- Cooney A, McCray PB Jr, and Sinn PL. Cystic Fibrosis Gene Therapy: Looking Back, Looking Forward. Genes 9: 538, 2018.
- Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nature Reviews Genetics 16: 45–56, 2014.
- Gibson RL, Burns JL, and Ramsey BW. Pathophysiology and Management of Pulmonary Infections in Cystic Fibrosis. American Journal of Respiratory and Critical Care Medicine 168: 918-951, 2003.
- Hisert KB, Heltshe SL, Pope C, Jorth P, Wu X, Edwards RM, Radey M, Accurso FJ, Wolter DJ, Cooke G, Adam RJ, Carter S, Grogan B, Launspach JL, Donnelly SC, Gallagher CG, Bruce JE, Stoltz DA, Welsh MJ, Hoffman LR, McKone EF, Singh PK. Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function Reduces Airway Bacteria and Inflammation in People with Cystic Fibrosis and Chronic Lung Infections. American Journal of Respiratory and Critical Care Medicine 195: 1617-1628, 2017.
- Lommatzsch ST, and Taylor-Cousar JL. The combination of tezacaftor and ivacaftor in the treatment of patients with cystic fibrosis: clinical evidence and future prospects in cystic fibrosis therapy. Therapeutic Advances in Respiratory Disease 13: 1-13, 2019.
- Marquette CR, and Luckie DB. Dissection of a Mechanistic Controversy in Cystic Fibrosis. JSM Genet Genomics 3(2): 1017, 2016.
- Ratjen FA. Cystic Fibrosis: Pathogenesis and Future Treatment Strategies. Respiratory Care 54: 595-605, 2009.
- Rey MM, Bonk MP, and Hadjiliadis D. Cystic Fibrosis: Emerging Understanding and Therapies. Annual Review of Medicine 70: 197-210, 2019.
- Yan Z, McCray PB Jr, and Engelhardt JF. Advances in gene therapy for cystic fibrosis lung disease. Human Molecular Genetics 28: 88-94, 2019.
Appendix
Figure 1- This is from reference 10. There are 6 classes of CF mutations. Class 1 mutations result in no CFTR protein made. Class II mutations result in CFTR protein that is inadequately processed. Class III mutations result in CFTR protein that is not regulated. Class IV mutations result in CFTR protein that has problems conducting the flow of chloride ions. Class V mutations result in CFTR protein that is partially defective. Class VI mutations result in CFTR protein that has accelerated degradation (9). Class I-III mutations result in severe lung disease and rapid breakdown of respiratory function. Class IV-VI mutations result in instability of the CFTR protein (3).
Figure 2- This is from reference 11. It depicts the molecular structure of the CFTR protein. The nucleotide binding domains (1 and 2) are shown, these are the sites of ATP binding. The R domain allows chloride ions to pass. The protein has intracellular and transmembrane portions.
Figure 3- This is from reference 1. Graph A shows the effect of inhaling hypertonic saline on the clearance of mucus in a patient with Cystic Fibrosis. The inhalation of HS resulted in an increase in basal mucus-ciliary clearance after two weeks of therapy when compared with measurements made before therapy was started. Graph B shows that this increase in the clearance of mucus was correlated with a significant increase in FEV1 over the same two-week interval.